Abstract
MSA2dist calculates pairwise distances between all sequences of a DNAStringSet or a AAStringSet using a custom score matrix and conducts codon based analysis. It uses scoring matrices to be used in these pairwise distance calcualtions which can be adapted to any scoring for DNA or AA characters. E.g. by using literal distances MSA2dist calcualtes pairwise IUPAC distances. DNAStringSet alignments can be analysed as codon alignments to look for synonymous and nonsynonymous substitutions in a parallelised fashion.
Introduction
Calculating pairwise distances of either DNA or AA sequences is a common task for evolutionary biologist. The distance calculations are either based on specific nucleotide, codon or amino acid models or on a scoring matrix.
Note: Sequences need to be pre-aligned into so
called multiple sequence alignments (MSA), which can be done with a
multitude of existing software. Just to mention for example mafft, muscle or the R
package msa.
The R package ape
for example offers the ape::dist.dna() function, which has
implemented a collection of different evolutionary models. MSA2dist
extends the possibility to directly calculate pairwise nucloetide
distances of an Biostrings::DNAStringSet object or pairwise
amino acid distances of an Biostrings::AAStringSet object.
The scoring matrix based calculations are implemented in
c++ with RcppThread to parallelise pairwise
combinations.
It is a non-trivial part to resolve haploid (1n) sequences from a
diploid (2n) individual (aka phasing) to further use the haploid
sequences for distance calculations. To cope with this situation,
MSA2dist uses a literal distance (Chang et al. 2017) which can be directly
applied on IUPAC nucleotide ambiguity encoded sequences
with the dnastring2dist() function. IUPAC
sequences can be for example obtained directly from mapped
BAM files and the angsd
-doFasta 4 option (Korneliussen,
Albrechtsen, and Nielsen 2014).
The Grantham’s score (Grantham 1974)
attempts to predict the distance between two amino acids, in an
evolutionary sense considering the amino acid composition, polarity and
molecular volume. MSA2dist offers with the
aastring2dist() function the possibility to obtain pairwise
distances of all sequences in an Biostrings::AAStringSet
(needs to be pre-aligned). The resulting distance matrix can be used to
calculate neighbor-joining trees via the R package ape.
Calculating synonymous (Ks) and nonsynonymous (Ka) substitutions from
coding sequences and its ratio Ka/Ks can be used as an indicator of
selective pressure acting on a protein. The
dnastring2kaks() function can be applied on pre-aligned
Biostrings::DNAStringSet() objects to calculate these
values either according to (Li 1993) via
the R package seqinr
or according to the model of (Nei and Gojobori
1986).
Further, all codons can be evaluated among the coding sequence
alignment and be plotted to for example protein domains with
substitutions or indels with the codonmat2xy()
function.
Installation
To install this package, start R (version “4.1”) and enter:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("MSA2dist")Sequence Format conversion
To be able to use distance calculation functions from other R
packages, like ape
or seqinr,
it is necessary to have dedicated sequence format conversion functions.
Here, some examples are shown, how to convert from and to a
Biostrings::DNAStringSet object.
?Biostrings::DNAStringSet() >>>
?seqinr::as.alignment()
## define two cds sequences
cds1 <- Biostrings::DNAString("ATGCAACATTGC")
cds2 <- Biostrings::DNAString("ATG---CATTGC")
cds1.cds2.aln <- c(Biostrings::DNAStringSet(cds1),
Biostrings::DNAStringSet(cds2))
## define names
names(cds1.cds2.aln) <- c("seq1", "seq2")
## convert into alignment
cds1.cds2.aln |> dnastring2aln()## $nb
## [1] 2
##
## $nam
## [1] "seq1" "seq2"
##
## $seq
## [1] "atgcaacattgc" "atg---cattgc"
##
## $com
## [1] NA
##
## attr(,"class")
## [1] "alignment"?seqinr::as.alignment() >>>
?Biostrings::DNAStringSet()
## convert back into DNAStringSet
cds1.cds2.aln |> dnastring2aln() |> aln2dnastring()## DNAStringSet object of length 2:
## width seq names
## [1] 12 ATGCAACATTGC seq1
## [2] 12 ATG---CATTGC seq2?Biostrings::DNAStringSet() >>>
?ape::DNAbin()
## convert into alignment
cds1.cds2.aln |> dnastring2dnabin()## 2 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 12
##
## Labels:
## seq1
## seq2
##
## Base composition:
## a c g t
## 0.286 0.238 0.190 0.286
## (Total: 24 bases)?ape::DNAbin() >>>
?Biostrings::DNAStringSet()
## convert back into DNAStringSet
cds1.cds2.aln |> dnastring2dnabin() |> dnabin2dnastring()## DNAStringSet object of length 2:
## width seq names
## [1] 12 ATGCAACATTGC seq1
## [2] 12 ATG---CATTGC seq2
## use woodmouse data
woodmouse |> dnabin2dnastring()## DNAStringSet object of length 15:
## width seq names
## [1] 965 NTTCGAAAAACACACCCACTACT...GCCCAATTACTCAGACCCTATA No305
## [2] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCTGTN No304
## [3] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCTATA No306
## [4] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAATACNNNN No0906S
## [5] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCNNNN No0908S
## ... ... ...
## [11] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCNNNN No1007S
## [12] 965 NNNNNNNNNNNNNNNNNNNNNNN...GCCCAATTACTCAAACCCNNNN No1114S
## [13] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCNNNN No1202S
## [14] 965 ATTCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCNNNN No1206S
## [15] 965 NNNCGAAAAACACACCCACTACT...GCCCAATTACTCAAACCCNNNN No1208S?Biostrings::AAStringSet() >>>
?seqinr::as.alignment()
## translate cds into aa
aa1.aa2.aln <- cds1.cds2.aln |> cds2aa()
## convert into alignment
aa1.aa2.aln |> aastring2aln()## $nb
## [1] 2
##
## $nam
## [1] "seq1" "seq2"
##
## $seq
## [1] "mqhc" "mxhc"
##
## $com
## [1] NA
##
## attr(,"class")
## [1] "alignment"?seqinr::as.alignment() >>>
?Biostrings::AAStringSet()
## convert back into AAStringSet
aa1.aa2.aln |> aastring2aln() |> aln2aastring()## AAStringSet object of length 2:
## width seq names
## [1] 4 MQHC seq1
## [2] 4 MXHC seq2?Biostrings::AAStringSet() >>>
?ape::as.AAbin()
## convert into AAbin
aa1.aa2.aln |> aastring2aabin()## 2 amino acid sequences in a list
##
## All sequences of the same length: 4?ape::as.AAbin() >>>
?Biostrings::AAStringSet()
## convert back into AAStringSet
aa1.aa2.aln |> aastring2aabin() |> aabin2aastring()## AAStringSet object of length 2:
## width seq names
## [1] 4 MQHC seq1
## [2] 4 MXHC seq2Frame aware Biostrings::DNAStringSet translation
(cds2aa())
To be able to translate a coding sequence into amino acids, sometimes
the sequences do not start at the first frame. The cds2aa
function can take an alternative codon start site into account
(frame = 1 or frame = 2 or
frame = 3). However, sometimes it is also necessary that
the resulting coding sequence length is a multiple of three. This can be
forced by using the shorten = TRUE option.
Simple translation:
## define two cds sequences
cds1 <- Biostrings::DNAString("ATGCAACATTGC")
cds2 <- Biostrings::DNAString("ATG---CATTGC")
cds1.cds2.aln <- c(Biostrings::DNAStringSet(cds1),
Biostrings::DNAStringSet(cds2))
## define names
names(cds1.cds2.aln) <- c("seq1", "seq2")
## translate cds into aa
cds1.cds2.aln |> cds2aa()## AAStringSet object of length 2:
## width seq names
## [1] 4 MQHC seq1
## [2] 4 MXHC seq2
aa1.aa2.aln <- cds1.cds2.aln |> cds2aa()Translation keeping multiple of three sequence length:
## translate cds into aa using frame = 2
## result is empty due to not multiple of three
cds1.cds2.aln |> cds2aa(frame=2)## AAStringSet object of length 0
## translate cds into aa using frame = 2 and shorten = TRUE
cds1.cds2.aln |> cds2aa(frame=2, shorten=TRUE)## AAStringSet object of length 2:
## width seq names
## [1] 3 CNI seq1
## [2] 3 XXI seq2
## translate cds into aa using frame = 3 and shorten = TRUE
cds1.cds2.aln |> cds2aa(frame=3, shorten=TRUE)## AAStringSet object of length 2:
## width seq names
## [1] 3 ATL seq1
## [2] 3 XXL seq2
## use woodmouse data
woodmouse |> dnabin2dnastring() |> cds2aa(shorten=TRUE)## AAStringSet object of length 15:
## width seq names
## [1] 321 XRKTHPLLKXISHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTL No305
## [2] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTL No304
## [3] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTL No306
## [4] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQIX No0906S
## [5] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No0908S
## ... ... ...
## [11] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No1007S
## [12] 321 XXXXXXXXXXXXXXXIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No1114S
## [13] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No1202S
## [14] 321 IRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No1206S
## [15] 321 XRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLIFRPITQTX No1208STranslation using alternative genetic code:
As you can see from the above example, the initial amino acids
I will change into M due to the mitochondrial
translation code and also some * stop codons will change
into a W amino acid.
## alternative genetic code
## use woodmouse data
woodmouse |> dnabin2dnastring() |> cds2aa(shorten=TRUE,
genetic.code=Biostrings::getGeneticCode("2"))## AAStringSet object of length 15:
## width seq names
## [1] 321 XRKTHPLLKXISHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTL No305
## [2] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTL No304
## [3] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTL No306
## [4] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQMX No0906S
## [5] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No0908S
## ... ... ...
## [11] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No1007S
## [12] 321 XXXXXXXXXXXXXXXIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No1114S
## [13] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No1202S
## [14] 321 MRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No1206S
## [15] 321 XRKTHPLLKIINHSFIDLPAPSN...LLPFLHTSKQRSLMFRPITQTX No1208SPairwise sequence comparison
Calculate pairwise AA distances (aastring2dist())
Grantham’s distance
## calculate pairwise AA distances based on Grantham's distance
aa.dist <- hiv |> cds2aa() |> aastring2dist(score=granthamMatrix())## Computing: [========================================] 100% (done)
## obtain distances
head(aa.dist$distSTRING)## U68496 U68497 U68498 U68499 U68500 U68501 U68502
## U68496 0.000000 4.43956 11.516484 9.879121 13.098901 17.05495 19.71429
## U68497 4.439560 0.00000 12.681319 10.406593 13.626374 18.21978 19.35165
## U68498 11.516484 12.68132 0.000000 8.769231 8.571429 13.25275 16.23077
## U68499 9.879121 10.40659 8.769231 0.000000 4.780220 15.58242 15.21978
## U68500 13.098901 13.62637 8.571429 4.780220 0.000000 15.38462 14.35165
## U68501 17.054945 18.21978 13.252747 15.582418 15.384615 0.00000 16.43956
## U68503 U68504 U68505 U68506 U68507 U68508
## U68496 17.82418 13.857143 14.24176 14.92308 18.01099 19.01099
## U68497 19.80220 13.890110 14.27473 14.82418 18.54945 18.90110
## U68498 16.15385 9.516484 10.27473 11.04396 12.15385 15.56044
## U68499 16.58242 10.483516 12.47253 12.01099 14.24176 13.52747
## U68500 14.95604 10.571429 12.56044 12.06593 13.84615 10.89011
## U68501 17.50549 10.758242 13.08791 11.78022 14.91209 13.86813
## obtain pairwise sites used
head(aa.dist$sitesUsed)## U68496 U68497 U68498 U68499 U68500 U68501 U68502 U68503 U68504 U68505
## U68496 91 91 91 91 91 91 91 91 91 91
## U68497 91 91 91 91 91 91 91 91 91 91
## U68498 91 91 91 91 91 91 91 91 91 91
## U68499 91 91 91 91 91 91 91 91 91 91
## U68500 91 91 91 91 91 91 91 91 91 91
## U68501 91 91 91 91 91 91 91 91 91 91
## U68506 U68507 U68508
## U68496 91 91 91
## U68497 91 91 91
## U68498 91 91 91
## U68499 91 91 91
## U68500 91 91 91
## U68501 91 91 91
## create and plot bionj tree
aa.dist.bionj <- ape::bionj(as.dist(aa.dist$distSTRING))
plot(aa.dist.bionj)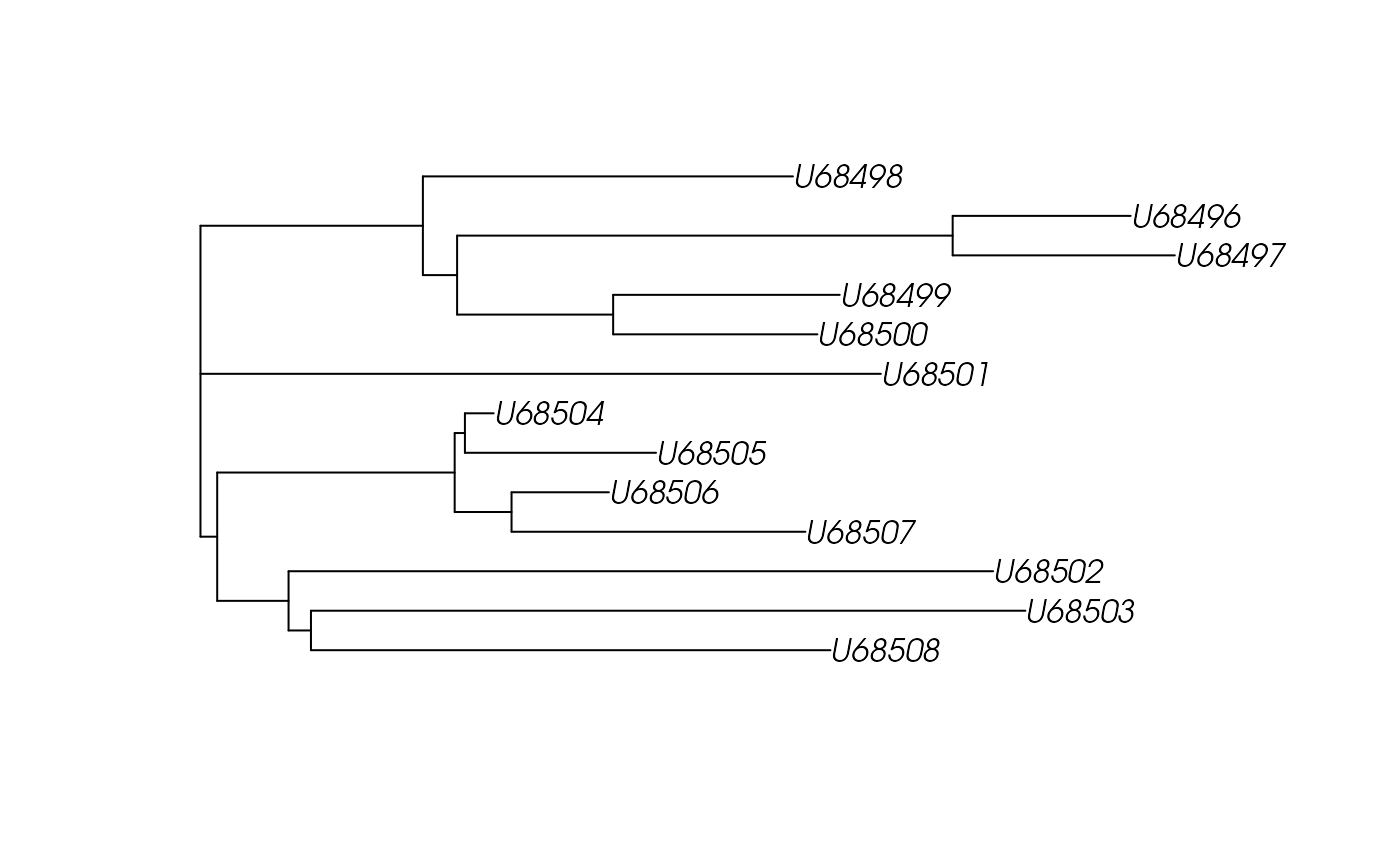
To use a different score matrix, here as an example the
AAMatrix from the R package alakazam
is used:
## use AAMatrix data
head(AAMatrix)## A B C D E F G H I J K L M N P Q R S T V W X Y Z * - .
## A 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 0 0
## B 1 0 1 1 1 1 1 1 1 1 1 1 1 0 1 1 0 1 1 1 1 0 1 1 1 0 0
## C 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 0 0
## D 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 0 0
## E 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 0 1 0 0
## F 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 0 0
aa.dist.AAMatrix <- hiv |> cds2aa() |> aastring2dist(score=AAMatrix)## Computing: [========================================] 100% (done)
head(aa.dist.AAMatrix$distSTRING)## U68496 U68497 U68498 U68499 U68500 U68501
## U68496 0.00000000 0.06593407 0.1758242 0.17582418 0.23076923 0.2417582
## U68497 0.06593407 0.00000000 0.2197802 0.19780220 0.25274725 0.2857143
## U68498 0.17582418 0.21978022 0.0000000 0.13186813 0.16483516 0.1868132
## U68499 0.17582418 0.19780220 0.1318681 0.00000000 0.08791209 0.2307692
## U68500 0.23076923 0.25274725 0.1648352 0.08791209 0.00000000 0.2637363
## U68501 0.24175824 0.28571429 0.1868132 0.23076923 0.26373626 0.0000000
## U68502 U68503 U68504 U68505 U68506 U68507 U68508
## U68496 0.2857143 0.2527473 0.2417582 0.2307692 0.2527473 0.2747253 0.3186813
## U68497 0.2967033 0.2747253 0.2527473 0.2417582 0.2637363 0.3076923 0.3186813
## U68498 0.2087912 0.2417582 0.1758242 0.1758242 0.2087912 0.1868132 0.2637363
## U68499 0.2197802 0.2417582 0.1758242 0.1758242 0.2087912 0.2087912 0.2197802
## U68500 0.2527473 0.2527473 0.2087912 0.2087912 0.2307692 0.2417582 0.2197802
## U68501 0.2527473 0.2637363 0.2087912 0.2307692 0.2197802 0.2417582 0.2747253Calculate pairwise DNA distances
(dnastring2dist())
ape::dist.dna models
## use hiv data
dna.dist <- hiv |> dnastring2dist(model="K80")## Computing: [========================================] 100% (done)
## obtain distances
head(dna.dist$distSTRING)## U68496 U68497 U68498 U68499 U68500 U68501
## U68496 0.00000000 0.03381189 0.07731910 0.08135801 0.11058044 0.1022214
## U68497 0.03381189 0.00000000 0.09396372 0.08960527 0.11916567 0.1194893
## U68498 0.07731910 0.09396372 0.00000000 0.05333488 0.07342797 0.0773191
## U68499 0.08135801 0.08960527 0.05333488 0.00000000 0.04143570 0.1022214
## U68500 0.11058044 0.11916567 0.07342797 0.04143570 0.00000000 0.1237391
## U68501 0.10222140 0.11948926 0.07731910 0.10222140 0.12373909 0.0000000
## U68502 U68503 U68504 U68505 U68506 U68507 U68508
## U68496 0.1503488 0.1282034 0.11948926 0.11991759 0.1157063 0.13779195 0.1684152
## U68497 0.1594427 0.1280155 0.13725343 0.12865927 0.1381014 0.15620300 0.1682063
## U68498 0.1105804 0.1067010 0.09823725 0.09004207 0.1110678 0.08987206 0.1366493
## U68499 0.1150839 0.1157063 0.09808005 0.09412740 0.1108796 0.10254762 0.1232278
## U68500 0.1457404 0.1286593 0.12357099 0.11948926 0.1323187 0.12415474 0.1234296
## U68501 0.1150839 0.1241547 0.10670096 0.10711515 0.1108796 0.11570627 0.1368233
## obtain pairwise sites used
head(dna.dist$sitesUsed)## U68496 U68497 U68498 U68499 U68500 U68501 U68502 U68503 U68504 U68505
## U68496 273 273 273 273 273 273 273 273 273 273
## U68497 273 273 273 273 273 273 273 273 273 273
## U68498 273 273 273 273 273 273 273 273 273 273
## U68499 273 273 273 273 273 273 273 273 273 273
## U68500 273 273 273 273 273 273 273 273 273 273
## U68501 273 273 273 273 273 273 273 273 273 273
## U68506 U68507 U68508
## U68496 273 273 273
## U68497 273 273 273
## U68498 273 273 273
## U68499 273 273 273
## U68500 273 273 273
## U68501 273 273 273It is also possible to compare the amino acid and nucleotide based trees:
## creation of the association matrix:
association <- cbind(aa.dist.bionj$tip.label, aa.dist.bionj$tip.label)
## cophyloplot
ape::cophyloplot(aa.dist.bionj,
dna.dist.bionj,
assoc=association,
length.line=4,
space=28,
gap=3,
rotate=FALSE)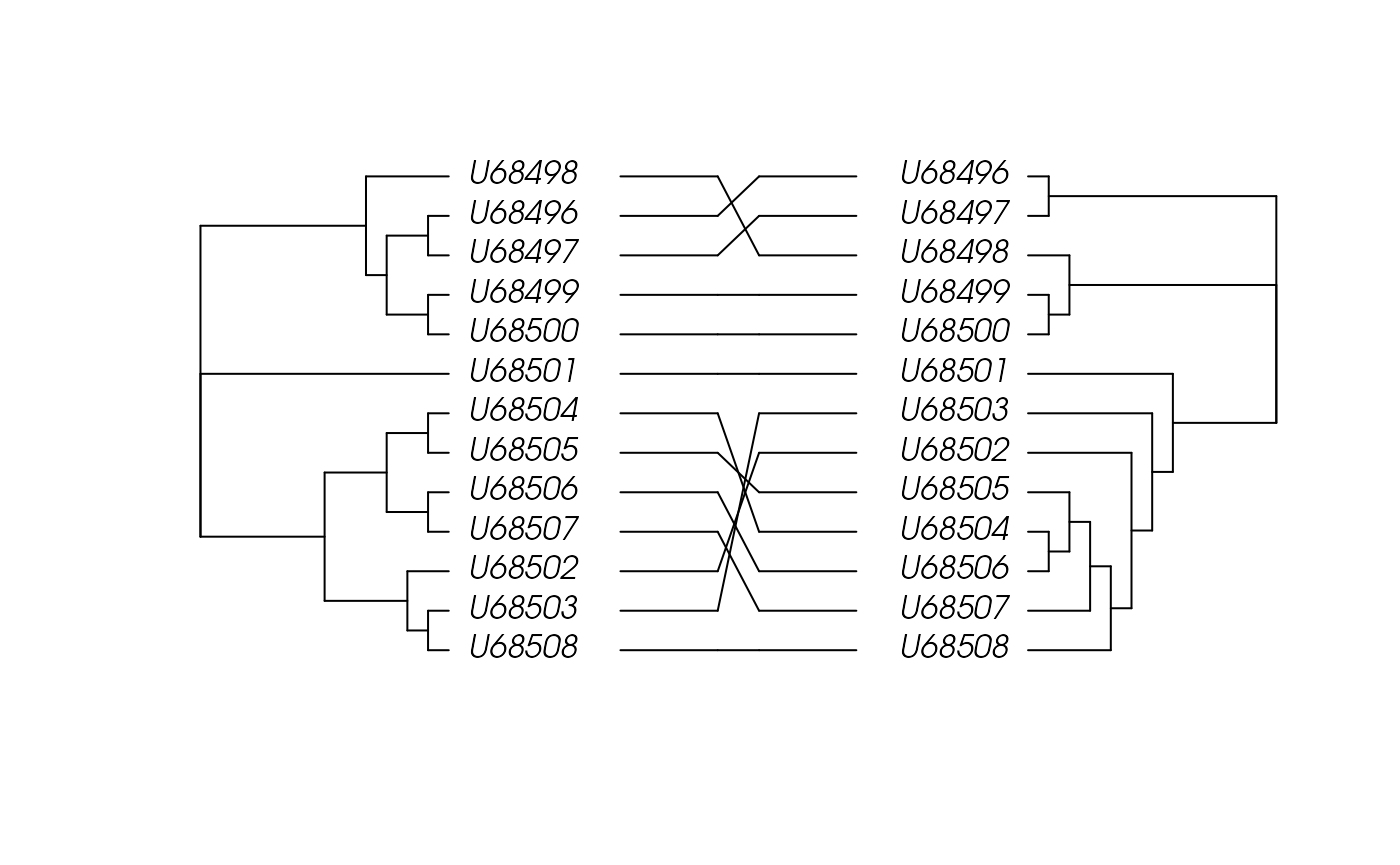
IUPAC distance
## use hiv data
hiv.dist.iupac <- head(hiv |> dnastring2dist(model="IUPAC"))## Computing: [========================================] 100% (done)
head(hiv.dist.iupac$distSTRING)## U68496 U68497 U68498 U68499 U68500 U68501
## U68496 0.00000000 0.03296703 0.07326007 0.07692308 0.10256410 0.09523810
## U68497 0.03296703 0.00000000 0.08791209 0.08424908 0.10989011 0.10989011
## U68498 0.07326007 0.08791209 0.00000000 0.05128205 0.06959707 0.07326007
## U68499 0.07692308 0.08424908 0.05128205 0.00000000 0.04029304 0.09523810
## U68500 0.10256410 0.10989011 0.06959707 0.04029304 0.00000000 0.11355311
## U68501 0.09523810 0.10989011 0.07326007 0.09523810 0.11355311 0.00000000
## U68502 U68503 U68504 U68505 U68506 U68507 U68508
## U68496 0.1355311 0.1172161 0.10989011 0.10989011 0.1062271 0.12454212 0.1501832
## U68497 0.1428571 0.1172161 0.12454212 0.11721612 0.1245421 0.13919414 0.1501832
## U68498 0.1025641 0.0989011 0.09157509 0.08424908 0.1025641 0.08424908 0.1245421
## U68499 0.1062271 0.1062271 0.09157509 0.08791209 0.1025641 0.09523810 0.1135531
## U68500 0.1318681 0.1172161 0.11355311 0.10989011 0.1208791 0.11355311 0.1135531
## U68501 0.1062271 0.1135531 0.09890110 0.09890110 0.1025641 0.10622711 0.1245421
## run multi-threaded
system.time(hiv |> dnastring2dist(model="IUPAC", threads=1))## Computing: [========================================] 100% (done)## user system elapsed
## 0.003 0.001 0.004
system.time(hiv |> dnastring2dist(model="IUPAC", threads=2))## Computing: [========================================] 100% (done)## user system elapsed
## 0.004 0.000 0.004Woodmouse data example:
## use woodmouse data
woodmouse.dist <- woodmouse |> dnabin2dnastring() |> dnastring2dist()## Computing: [========================================] 100% (done)
head(woodmouse.dist$distSTRING)## No305 No304 No306 No0906S No0908S No0909S
## No305 0.00000000 0.016684046 0.013541667 0.018789144 0.01670146 0.01670146
## No304 0.01668405 0.000000000 0.005208333 0.013555787 0.01147028 0.01564129
## No306 0.01354167 0.005208333 0.000000000 0.009384776 0.00729927 0.01147028
## No0906S 0.01878914 0.013555787 0.009384776 0.000000000 0.01248699 0.01664932
## No0908S 0.01670146 0.011470282 0.007299270 0.012486993 0.00000000 0.01456816
## No0909S 0.01670146 0.015641293 0.011470282 0.016649324 0.01456816 0.00000000
## No0910S No0912S No0913S No1103S No1007S No1114S
## No305 0.017745303 0.014613779 0.018789144 0.012526096 0.016701461 0.01531729
## No304 0.012513034 0.013555787 0.005213764 0.011470282 0.015641293 0.01642935
## No306 0.008342023 0.009384776 0.005213764 0.007299270 0.011470282 0.01533406
## No0906S 0.009365245 0.014568158 0.012486993 0.012486993 0.016649324 0.02076503
## No0908S 0.011446410 0.012486993 0.012486993 0.010405827 0.014568158 0.02076503
## No0909S 0.015608741 0.010405827 0.016649324 0.008324662 0.002081165 0.02076503
## No1202S No1206S No1208S
## No305 0.016701461 0.016701461 0.018828452
## No304 0.011470282 0.012513034 0.017782427
## No306 0.007299270 0.008342023 0.013598326
## No0906S 0.008324662 0.011446410 0.018789144
## No0908S 0.010405827 0.009365245 0.016701461
## No0909S 0.014568158 0.015608741 0.002087683Coding sequences
Calculating synonymous and nonsynonymous substitutions
(dnastring2kaks())
## use hiv data
## model Li
head(hiv |> dnastring2kaks(model="Li"))## Joining with `by = join_by(seq1, seq2)`
## Joining with `by = join_by(seq1, seq2)`
## Joining with `by = join_by(seq1, seq2)`## Comp1 Comp2 seq1 seq2 ka ks vka vks
## 1 1 2 U68496 U68497 0.03026357 0.03170319 0.0003051202 0.0004007730
## 2 1 3 U68496 U68498 0.09777332 0.01761416 0.0009314173 0.0005091970
## 3 1 4 U68496 U68499 0.10295875 0.01767311 0.0009595527 0.0005787387
## 4 1 5 U68496 U68500 0.13461355 0.04639690 0.0013731885 0.0020497481
## 5 1 6 U68496 U68501 0.12607831 0.02844294 0.0013277282 0.0006310195
## 6 1 7 U68496 U68502 0.17441037 0.10926532 0.0017871934 0.0040766687
## Ka Ks Ka/Ks
## 1 0.03026357 0.03170319 0.9545907
## 2 0.09777332 0.01761416 5.5508365
## 3 0.10295875 0.01767311 5.8257302
## 4 0.13461355 0.04639690 2.9013480
## 5 0.12607831 0.02844294 4.4326750
## 6 0.17441037 0.10926532 1.5962097
## model NG86
head(hiv |> dnastring2kaks(model="NG86", threads=1))## Comp1 Comp2 seq1 seq2 Codons Compared Ambigiuous Indels Ns
## result.1 1 2 U68496 U68497 91 91 0 0 0
## result.2 1 3 U68496 U68498 91 91 0 0 0
## result.3 1 4 U68496 U68499 91 91 0 0 0
## result.4 1 5 U68496 U68500 91 91 0 0 0
## result.5 1 6 U68496 U68501 91 91 0 0 0
## result.6 1 7 U68496 U68502 91 91 0 0 0
## Sd Sn S N
## result.1 3 6 57 216
## result.2 1.5 18.5 57.5 215.5
## result.3 1.5 19.5 56.5 216.5
## result.4 2.5 25.5 56.1666666666667 216.833333333333
## result.5 2.5 23.5 57.3333333333333 215.666666666667
## result.6 5.83333333333333 31.1666666666667 57.5 215.5
## ps pn pn/ps
## result.1 0.0526315789473684 0.0277777777777778 0.527777777777778
## result.2 0.0260869565217391 0.08584686774942 3.2907965970611
## result.3 0.0265486725663717 0.0900692840646651 3.39260969976905
## result.4 0.0445103857566766 0.117601844734819 2.64212144504228
## result.5 0.0436046511627907 0.108964451313756 2.49891808346213
## result.6 0.101449275362319 0.144624903325599 1.42558833278091
## ds dn dn/ds
## result.1 0.0545695157118212 0.0283052459871352 0.518700699793888
## result.2 0.026551445288187 0.0911703470876381 3.43372445823884
## result.3 0.0270299523623977 0.0959537646568416 3.54990505977829
## result.4 0.0458858673563109 0.127915513677956 2.78768869474935
## result.5 0.0449236061858017 0.117741219733924 2.62092093067847
## result.6 0.108999740568461 0.160668709585171 1.47402836692312
## Ka Ks Ka/Ks
## result.1 0.0283052459871352 0.0545695157118212 0.518700699793888
## result.2 0.0911703470876381 0.026551445288187 3.43372445823884
## result.3 0.0959537646568416 0.0270299523623977 3.54990505977829
## result.4 0.127915513677956 0.0458858673563109 2.78768869474935
## result.5 0.117741219733924 0.0449236061858017 2.62092093067847
## result.6 0.160668709585171 0.108999740568461 1.47402836692312Using any model from KaKs_Calculator 2.0
models to choose from KaKs_Calculator 2.0 (D. Wang et al. 2010) are:
- MS (D. Wang et al. 2010)
- MA (D. Wang et al. 2010)
- GNG (D. Wang et al. 2010)
- GLWL (D. Wang et al. 2010)
- GLPB (D. Wang et al. 2010)
- GMLWL (D. Wang et al. 2010)
- GMLPB (D. Wang et al. 2010)
- GYN (D. Wang et al. 2010)
- LWL (Li, Wu, and Luo 1985)
- LPB Pamilo and Bianchi (1993)
- NG (Nei and Gojobori 1986)
- MLPB (Tzeng, Pan, and Li 2004)
- GY (Goldman and Yang 1994)
- YN (Yang et al. 2000)
- MYN D.-P. Wang et al. (2009)
## use hiv data
## model MYN
head(hiv |> dnastring2kaks(model="MYN"))## Comp1 Comp2 seq1 seq2 Method Ka Ks Ka/Ks
## U68496_U68497 1 2 U68496 U68497 MYN 0.0259184 0.0969684 0.267287
## U68496_U68498 1 3 U68496 U68498 MYN 0.0824755 0.0443815 1.85833
## U68496_U68499 1 4 U68496 U68499 MYN 0.0859047 0.051651 1.66318
## U68496_U68500 1 5 U68496 U68500 MYN 0.112508 0.112329 1.00159
## U68496_U68501 1 6 U68496 U68501 MYN 0.106234 0.0783914 1.35518
## U68496_U68502 1 7 U68496 U68502 MYN 0.147219 0.177543 0.829206
## P-Value(Fisher) Length S-Sites N-Sites Fold-Sites(0:2:4)
## U68496_U68497 0.104824 273 36.5412 236.459 NA
## U68496_U68498 0.665602 273 34.4592 238.541 NA
## U68496_U68499 0.645824 273 31.032 241.968 NA
## U68496_U68500 0.96729 273 29.8065 243.193 NA
## U68496_U68501 0.731834 273 34.8366 238.163 NA
## U68496_U68502 0.600487 273 36.4985 236.501 NA
## Substitutions S-Substitutions N-Substitutions
## U68496_U68497 9 3 6
## U68496_U68498 20 1.4506 18.5494
## U68496_U68499 21 1.46511 19.5349
## U68496_U68500 28 2.74801 25.252
## U68496_U68501 26 2.53653 23.4635
## U68496_U68502 37 5.69633 31.3037
## Fold-S-Substitutions(0:2:4) Fold-N-Substitutions(0:2:4)
## U68496_U68497 NA NA
## U68496_U68498 NA NA
## U68496_U68499 NA NA
## U68496_U68500 NA NA
## U68496_U68501 NA NA
## U68496_U68502 NA NA
## Divergence-Time
## U68496_U68497 0.0354285
## U68496_U68498 0.0776671
## U68496_U68499 0.0820111
## U68496_U68500 0.112488
## U68496_U68501 0.102681
## U68496_U68502 0.151273
## Substitution-Rate-Ratio(rTC:rAG:rTA:rCG:rTG:rCA/rCA)
## U68496_U68497 2:2:1:1:1:1
## U68496_U68498 2.33502:2.98568:1:1:1:1
## U68496_U68499 1.29734:3.77147:1:1:1:1
## U68496_U68500 0.919154:2.95998:1:1:1:1
## U68496_U68501 1.57937:2.80997:1:1:1:1
## U68496_U68502 1.38699:4.11551:1:1:1:1
## GC(1:2:3) ML-Score AICc Akaike-Weight
## U68496_U68497 0.300366(0.362637:0.368132:0.17033) NA NA NA
## U68496_U68498 0.305861(0.373626:0.373626:0.17033) NA NA NA
## U68496_U68499 0.304029(0.373626:0.362637:0.175824) NA NA NA
## U68496_U68500 0.300366(0.362637:0.362637:0.175824) NA NA NA
## U68496_U68501 0.302198(0.362637:0.362637:0.181319) NA NA NA
## U68496_U68502 0.320513(0.368132:0.384615:0.208791) NA NA NA
## Model
## U68496_U68497 NA
## U68496_U68498 NA
## U68496_U68499 NA
## U68496_U68500 NA
## U68496_U68501 NA
## U68496_U68502 NA
## model YN
head(hiv |> dnastring2kaks(model="YN", threads=1))## Comp1 Comp2 seq1 seq2 Method Ka Ks Ka/Ks
## U68496_U68497 1 2 U68496 U68497 YN 0.0259337 0.0848079 0.305794
## U68496_U68498 1 3 U68496 U68498 YN 0.0833439 0.0402529 2.07051
## U68496_U68499 1 4 U68496 U68499 YN 0.0888328 0.0375284 2.36709
## U68496_U68500 1 5 U68496 U68500 YN 0.11672 0.074978 1.55673
## U68496_U68501 1 6 U68496 U68501 YN 0.108488 0.0673923 1.60979
## U68496_U68502 1 7 U68496 U68502 YN 0.153774 0.135014 1.13894
## P-Value(Fisher) Length S-Sites N-Sites Fold-Sites(0:2:4)
## U68496_U68497 0.110988 273 37.4117 235.588 NA
## U68496_U68498 0.441303 273 36.6562 236.344 NA
## U68496_U68499 0.471701 273 38.0351 234.965 NA
## U68496_U68500 0.547288 273 37.4853 235.515 NA
## U68496_U68501 0.540854 273 39.4297 233.57 NA
## U68496_U68502 0.811025 273 44.966 228.034 NA
## Substitutions S-Substitutions N-Substitutions
## U68496_U68497 9 3 6
## U68496_U68498 20 1.42507 18.5749
## U68496_U68499 21 1.38331 19.6167
## U68496_U68500 28 2.62481 25.3752
## U68496_U68501 26 2.49647 23.5035
## U68496_U68502 37 5.51119 31.4888
## Fold-S-Substitutions(0:2:4) Fold-N-Substitutions(0:2:4)
## U68496_U68497 NA NA
## U68496_U68498 NA NA
## U68496_U68499 NA NA
## U68496_U68500 NA NA
## U68496_U68501 NA NA
## U68496_U68502 NA NA
## Divergence-Time
## U68496_U68497 0.0340018
## U68496_U68498 0.077558
## U68496_U68499 0.081685
## U68496_U68500 0.110989
## U68496_U68501 0.102552
## U68496_U68502 0.150684
## Substitution-Rate-Ratio(rTC:rAG:rTA:rCG:rTG:rCA/rCA)
## U68496_U68497 2.90975:2.90975:1:1:1:1
## U68496_U68498 2.92242:2.92242:1:1:1:1
## U68496_U68499 3.24306:3.24306:1:1:1:1
## U68496_U68500 2.4231:2.4231:1:1:1:1
## U68496_U68501 2.49234:2.49234:1:1:1:1
## U68496_U68502 3.27339:3.27339:1:1:1:1
## GC(1:2:3) ML-Score AICc Akaike-Weight
## U68496_U68497 0.300366(0.362637:0.368132:0.17033) NA NA NA
## U68496_U68498 0.305861(0.373626:0.373626:0.17033) NA NA NA
## U68496_U68499 0.304029(0.373626:0.362637:0.175824) NA NA NA
## U68496_U68500 0.300366(0.362637:0.362637:0.175824) NA NA NA
## U68496_U68501 0.302198(0.362637:0.362637:0.181319) NA NA NA
## U68496_U68502 0.320513(0.368132:0.384615:0.208791) NA NA NA
## Model
## U68496_U68497 NA
## U68496_U68498 NA
## U68496_U68499 NA
## U68496_U68500 NA
## U68496_U68501 NA
## U68496_U68502 NAUsing indices to calculate Ka/Ks
## use hiv data
idx <- list(c(2, 3), c(5,7,9))
## model MYN
head(hiv |> indices2kaks(idx, model="MYN"))## Comp1 Comp2 seq1 seq2 Method Ka Ks Ka/Ks
## U68497_U68498 2 3 U68497 U68498 MYN 0.101709 0.0408904 2.48736
## U68500_U68502 5 7 U68500 U68502 MYN 0.138575 0.219015 0.632718
## U68500_U68504 5 9 U68500 U68504 MYN 0.110801 0.230614 0.480461
## U68502_U68504 7 9 U68502 U68504 MYN 0.0973798 0.259293 0.375559
## P-Value(Fisher) Length S-Sites N-Sites Fold-Sites(0:2:4)
## U68497_U68498 0.455523 273 31.8006 241.199 NA
## U68500_U68502 0.276773 273 35.1942 237.806 NA
## U68500_U68504 0.144952 273 36.342 236.658 NA
## U68502_U68504 0.0262893 273 36.862 236.138 NA
## Substitutions S-Substitutions N-Substitutions
## U68497_U68498 24 1.21809 22.7819
## U68500_U68502 36 6.19865 29.8013
## U68500_U68504 31 6.8704 24.1296
## U68502_U68504 29 7.5258 21.4742
## Fold-S-Substitutions(0:2:4) Fold-N-Substitutions(0:2:4)
## U68497_U68498 NA NA
## U68500_U68502 NA NA
## U68500_U68504 NA NA
## U68502_U68504 NA NA
## Divergence-Time
## U68497_U68498 0.0946247
## U68500_U68502 0.148945
## U68500_U68504 0.126751
## U68502_U68504 0.119242
## Substitution-Rate-Ratio(rTC:rAG:rTA:rCG:rTG:rCA/rCA)
## U68497_U68498 2.32976:4.35875:1:1:1:1
## U68500_U68502 0.79544:3.88533:1:1:1:1
## U68500_U68504 8.06492:8.71435:1:1:1:1
## U68502_U68504 2.03541:4.21617:1:1:1:1
## GC(1:2:3) ML-Score AICc Akaike-Weight
## U68497_U68498 0.309524(0.373626:0.379121:0.175824) NA NA NA
## U68500_U68502 0.324176(0.368132:0.384615:0.21978) NA NA NA
## U68500_U68504 0.300366(0.362637:0.379121:0.159341) NA NA NA
## U68502_U68504 0.320513(0.368132:0.401099:0.192308) NA NA NA
## Model
## U68497_U68498 NA
## U68500_U68502 NA
## U68500_U68504 NA
## U68502_U68504 NA
## model YN
head(hiv |> indices2kaks(idx, model="YN", threads=1))## Comp1 Comp2 seq1 seq2 Method Ka Ks Ka/Ks
## U68497_U68498 2 3 U68497 U68498 YN 0.104384 0.0325135 3.21047
## U68500_U68502 5 7 U68500 U68502 YN 0.146272 0.145608 1.00456
## U68500_U68504 5 9 U68500 U68504 YN 0.109735 0.229011 0.479167
## U68502_U68504 7 9 U68502 U68504 YN 0.100536 0.20586 0.488369
## P-Value(Fisher) Length S-Sites N-Sites Fold-Sites(0:2:4)
## U68497_U68498 0.19413 273 37.4724 235.528 NA
## U68500_U68502 0.99913 273 45.3387 227.661 NA
## U68500_U68504 0.140659 273 35.6546 237.345 NA
## U68502_U68504 0.0951223 273 43.0639 229.936 NA
## Substitutions S-Substitutions N-Substitutions
## U68497_U68498 24 1.18051 22.8195
## U68500_U68502 36 5.89752 30.1025
## U68500_U68504 31 6.85493 24.1451
## U68502_U68504 29 7.40851 21.5915
## Fold-S-Substitutions(0:2:4) Fold-N-Substitutions(0:2:4)
## U68497_U68498 NA NA
## U68500_U68502 NA NA
## U68500_U68504 NA NA
## U68502_U68504 NA NA
## Divergence-Time
## U68497_U68498 0.0945188
## U68500_U68502 0.146162
## U68500_U68504 0.125312
## U68502_U68504 0.11715
## Substitution-Rate-Ratio(rTC:rAG:rTA:rCG:rTG:rCA/rCA)
## U68497_U68498 3.99769:3.99769:1:1:1:1
## U68500_U68502 3.02484:3.02484:1:1:1:1
## U68500_U68504 3.14439:3.14439:1:1:1:1
## U68502_U68504 3.568:3.568:1:1:1:1
## GC(1:2:3) ML-Score AICc Akaike-Weight
## U68497_U68498 0.309524(0.373626:0.379121:0.175824) NA NA NA
## U68500_U68502 0.324176(0.368132:0.384615:0.21978) NA NA NA
## U68500_U68504 0.300366(0.362637:0.379121:0.159341) NA NA NA
## U68502_U68504 0.320513(0.368132:0.401099:0.192308) NA NA NA
## Model
## U68497_U68498 NA
## U68500_U68502 NA
## U68500_U68504 NA
## U68502_U68504 NACodon comparison
As an example for the codon comparison data from the Human Immunodeficiency Virus Type 1 is used (Ganeshan et al. 1997), (Yang et al. 2000).
The window plots are constructed with the R package ggplot2.
Create codon matrix (dnastring2codonmat())
## define two cds sequences
cds1 <- Biostrings::DNAString("ATGCAACATTGC")
cds2 <- Biostrings::DNAString("ATG---CATTGC")
cds1.cds2.aln <- c(Biostrings::DNAStringSet(cds1),
Biostrings::DNAStringSet(cds2))
## convert into alignment
cds1.cds2.aln |> dnastring2codonmat()## [,1] [,2]
## [1,] "ATG" "ATG"
## [2,] "CAA" "---"
## [3,] "CAT" "CAT"
## [4,] "TGC" "TGC"Like the cds2aa() function, also the
dnastring2codonmat() function is implemented to handle
different frames.
## use frame 2 and shorten to circumvent multiple of three error
cds1 <- Biostrings::DNAString("-ATGCAACATTGC-")
cds2 <- Biostrings::DNAString("-ATG---CATTGC-")
cds1.cds2.aln <- c(Biostrings::DNAStringSet(cds1),
Biostrings::DNAStringSet(cds2))
cds1.cds2.aln |> dnastring2codonmat(frame=2, shorten=TRUE)## [,1] [,2]
## [1,] "ATG" "ATG"
## [2,] "CAA" "---"
## [3,] "CAT" "CAT"
## [4,] "TGC" "TGC"Calculate average behavior of each codon
(codonmat2xy())
## use hiv data
## calculate average behavior
hiv.xy <- hiv |> dnastring2codonmat() |> codonmat2xy()## Joining with `by = join_by(Codon)`
## Joining with `by = join_by(Codon)`
## Joining with `by = join_by(Codon)`Plot average behavior of each codon
print(hiv.xy |> dplyr::select(Codon,SynMean,NonSynMean,IndelMean) |>
tidyr::gather(variable, values, -Codon) |>
ggplot2::ggplot(aes(x=Codon, y=values)) +
ggplot2::geom_line(aes(colour=factor(variable))) +
ggplot2::geom_point(aes(colour=factor(variable))) +
ggplot2::ggtitle("HIV-1 sample 136 patient 1 from
Sweden envelope glycoprotein (env) gene"))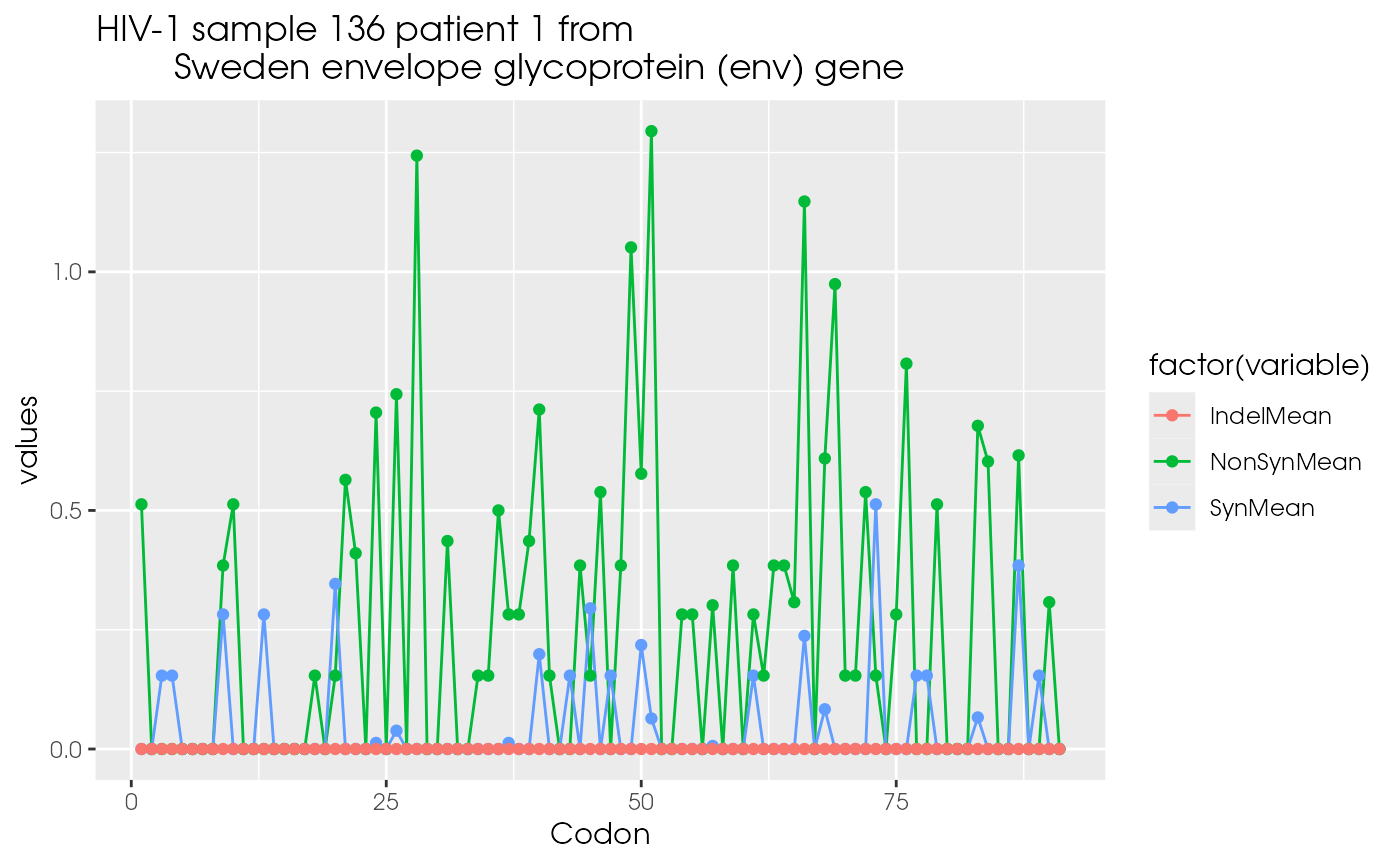
References
Session Info
## R version 4.3.3 (2024-02-29)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Debian GNU/Linux trixie/sid
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] ggplot2_3.5.0 tidyr_1.3.1 dplyr_1.1.4
## [4] ape_5.7-1 Biostrings_2.70.3 GenomeInfoDb_1.38.8
## [7] XVector_0.42.0 IRanges_2.36.0 S4Vectors_0.40.2
## [10] BiocGenerics_0.48.1 MSA2dist_1.7.5 BiocStyle_2.30.0
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.4 xfun_0.43 bslib_0.6.2
## [4] lattice_0.22-6 tzdb_0.4.0 vctrs_0.6.5
## [7] tools_4.3.3 bitops_1.0-7 generics_0.1.3
## [10] parallel_4.3.3 tibble_3.2.1 fansi_1.0.6
## [13] highr_0.10 pkgconfig_2.0.3 desc_1.4.3
## [16] lifecycle_1.0.4 GenomeInfoDbData_1.2.11 farver_2.1.1
## [19] stringr_1.5.1 compiler_4.3.3 textshaping_0.3.7
## [22] munsell_0.5.0 codetools_0.2-19 htmltools_0.5.8
## [25] sass_0.4.9 RCurl_1.98-1.14 yaml_2.3.8
## [28] pkgdown_2.0.7 pillar_1.9.0 crayon_1.5.2
## [31] jquerylib_0.1.4 seqinr_4.2-36 MASS_7.3-60.0.1
## [34] cachem_1.0.8 iterators_1.0.14 foreach_1.5.2
## [37] nlme_3.1-164 tidyselect_1.2.1 digest_0.6.35
## [40] stringi_1.8.3 purrr_1.0.2 bookdown_0.38
## [43] labeling_0.4.3 ade4_1.7-22 fastmap_1.1.1
## [46] grid_4.3.3 colorspace_2.1-0 cli_3.6.2
## [49] magrittr_2.0.3 utf8_1.2.4 withr_3.0.0
## [52] readr_2.1.5 scales_1.3.0 rmarkdown_2.26
## [55] ragg_1.2.7 hms_1.1.3 memoise_2.0.1
## [58] evaluate_0.23 knitr_1.45 GenomicRanges_1.54.1
## [61] doParallel_1.0.17 rlang_1.1.3 Rcpp_1.0.12
## [64] glue_1.7.0 BiocManager_1.30.22 jsonlite_1.8.8
## [67] R6_2.5.1 systemfonts_1.0.5 fs_1.6.3
## [70] zlibbioc_1.48.2